
l'editoriale
Cerca
MALATTIE RARE

17 Novembre 2024 - 21:45

La malattia di Charcot-Marie-Tooth (CMT) è una delle neuropatie ereditarie più comuni al mondo, con una prevalenza di circa 1 persona su 2.500. Caratterizzata dal danneggiamento progressivo dei nervi che controllano i movimenti muscolari e la sensibilità, questa malattia colpisce prevalentemente la parte inferiore delle gambe, causando debolezza muscolare, atrofia e perdita di sensazione.
Cos’è la malattia di Charcot-Marie-Tooth?
La malattia di Charcot-Marie-Tooth è una condizione neurologica che interessa sia i nervi motori (che controllano il movimento dei muscoli) che i nervi sensoriali (che trasmettono le informazioni sensoriali al cervello). Sebbene la malattia possa esordire in qualsiasi momento della vita, generalmente si manifesta durante l'infanzia o l'adolescenza.
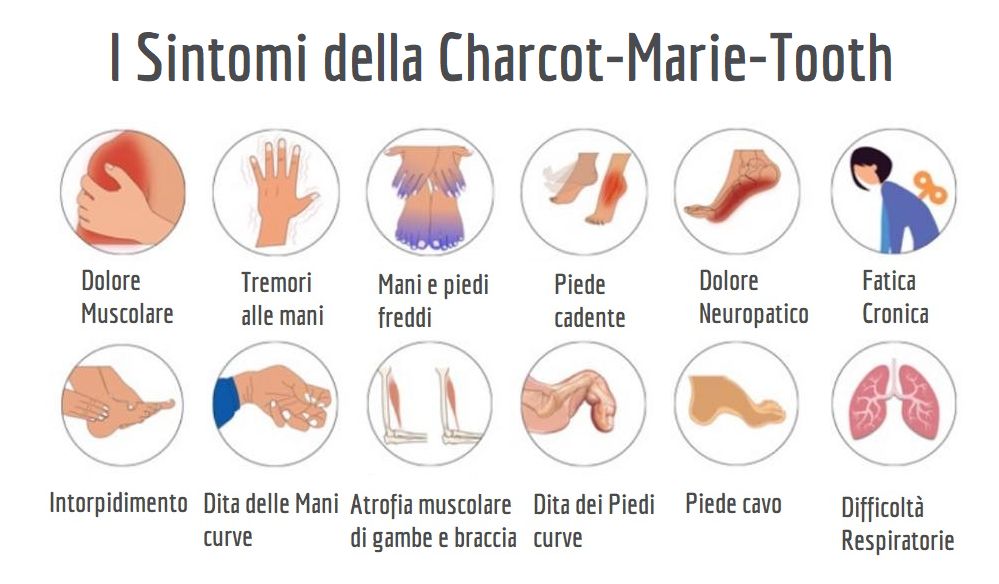
I sintomi iniziano solitamente nella parte inferiore delle gambe, con un progressivo indebolimento dei muscoli, che può estendersi alle cosce e, talvolta, anche alle mani. Questo porta a difficoltà nei movimenti, come l'incapacità di sollevare la parte anteriore del piede, causando il cosiddetto "piede cadente". L'atrofia muscolare riduce anche il volume dei muscoli delle gambe, portando a una deformità simile alla "gamba a cicogna", con il piede che si inclina in modo anomalo. A questi sintomi si aggiunge la perdita di sensibilità, con la difficoltà di percepire vibrazioni, dolore e temperatura, che peggiora progressivamente lungo gli arti.

Diagnosi e gestione della malattia
Per diagnosticare la CMT, i medici effettuano test diagnostici specifici come l’elettromiografia (EMG) e gli studi di conduzione nervosa, che permettono di analizzare la velocità di trasmissione degli impulsi lungo i nervi danneggiati. Sebbene non esista un trattamento in grado di fermare la progressione della malattia, interventi di fisioterapia, terapia occupazionale e l'uso di supporti ortopedici (come tutori per caviglie e piedi) possono migliorare la qualità della vita dei pazienti, aiutandoli a mantenere una certa funzionalità motoria.
Le diverse forme di Charcot-Marie-Tooth
La malattia di Charcot-Marie-Tooth non è una condizione unica, ma comprende una serie di varianti, classificate in base al tipo di danno che provoca. Le due principali categorie di CMT sono:
Demielinizzazione: In questa forma, la guaina mielinica che ricopre gli assoni (le fibre nervose che trasmettono gli impulsi) viene danneggiata o distrutta. La mielina agisce come un "isolante" per i nervi, facilitando la rapida trasmissione degli impulsi nervosi. Quando la mielina viene danneggiata, la conduzione nervosa rallenta, causando debolezza e perdita di sensibilità.
Danno all'assone: In altre forme della CMT, il danno colpisce direttamente l'assone, la parte del nervo che invia i segnali. In alcuni casi, l'assone può morire, impedendo così la trasmissione dei segnali nervosi.
La malattia può essere ereditata in modi diversi a seconda del tipo genetico. La forma autosomica dominante è la più comune, il che significa che una sola copia del gene mutato (ereditato da uno dei due genitori) è sufficiente per causare la malattia. Altre forme di CMT possono essere autosomiche recessive (quando è necessario ereditare il gene mutato da entrambi i genitori) o legate al sesso (nel caso in cui il gene mutato si trovi sul cromosoma X).
Sintomi e progressione della malattia
I sintomi della CMT possono variare notevolmente da persona a persona, ma di solito cominciano a manifestarsi durante l'infanzia o l'adolescenza. Nei casi più gravi, la debolezza muscolare progredisce rapidamente, causando difficoltà motorie e compromissione della mobilità. Al contrario, nelle forme più lievi, i sintomi possono essere più contenuti e limitarsi a problemi come il piede cavo o le dita a martello, un difetto dell’allineamento delle dita dei piedi.
Nelle forme più severe, i muscoli delle gambe e delle mani possono ridursi significativamente di volume, e la perdita di sensibilità diventa sempre più estesa, rendendo difficoltosa anche l’esecuzione di attività quotidiane. Tuttavia, la progressione della malattia è generalmente lenta e non incide sull’aspettativa di vita. La malattia di Charcot-Marie-Tooth non compromette infatti le funzioni vitali come la respirazione o il cuore.
Una forma rara di neuropatia che presenta sintomi simili alla CMT è la sindrome di Dejerine-Sottas (nota anche come neuropatia interstiziale ipertrofica). Questo disturbo, sebbene condivida con la CMT la perdita di sensibilità e i problemi motori, causa un peggioramento più rapido dei sintomi. La sindrome di Dejerine-Sottas inizia generalmente nell’infanzia, e porta alla perdita precoce dei riflessi e della sensibilità. Anche questa forma può essere ereditaria, con modalità di trasmissione simili a quelle della CMT.
Attualmente, non esistono trattamenti farmacologici in grado di fermare o curare la malattia di Charcot-Marie-Tooth. Tuttavia, la ricerca scientifica è in continuo progresso, e le nuove terapie genetiche e cellulari potrebbero portare a trattamenti in grado di modificare l’evoluzione della malattia in futuro. Nel frattempo, le persone affette da CMT possono migliorare la loro qualità della vita con un adeguato supporto terapeutico e una gestione preventiva della malattia, cercando di mantenere l’autonomia il più a lungo possibile. Le strategie di supporto includono l'uso di protesi, ortesi e la fisioterapia, che possono ridurre il dolore e migliorare la funzionalità dei muscoli e delle articolazioni.
|
|
|
I più letti
 L'associazione aderisce all'Istituto dell'Autodisciplina Pubblicitaria - IAP vincolando tutti i suoi Associati al rispetto del Codice di Autodisciplina della Comunicazione Commerciale e delle decisioni del Giurì e de Comitato di Controllo.
L'associazione aderisce all'Istituto dell'Autodisciplina Pubblicitaria - IAP vincolando tutti i suoi Associati al rispetto del Codice di Autodisciplina della Comunicazione Commerciale e delle decisioni del Giurì e de Comitato di Controllo.
CronacaQui.it | Direttore responsabile: Andrea Monticone
Vicedirettore: Marco Bardesono Capo servizio cronaca: Claudio Neve
Editore: Editoriale Argo s.r.l. Via Principe Tommaso 30 – 10125 Torino | C.F.08313560016 | P.IVA.08313560016. Redazione Torino: via Principe Tommaso, 30 – 10125 Torino |Tel. 011.6669, Email redazione@torinocronaca.it. Fax. 0116669232 ISSN 2611-2272 Amministratore unico e responsabile trattamento dati e sicurezza: Walter Altea
Registrazione tribunale n° 1877 del 14.03.1950 Tribunale di Milano
La società percepisce i contributi di cui al decreto legislativo 15 maggio 2017, n. 70. Indicazione resa ai sensi della lettera f) del comma 2 dell’articolo 5 del medesimo decreto legislativo..


 Edizione
Edizione



")





 scuote Torino, alla ricerca di un rilancio dell'auto")

